Concepto. El término glomerulopatía (GP) designa un conjunto de enfermedades que se caracterizan por una pérdida de las funciones normales del glomérulo renal. Se caracterizan por la aparición de elementos formes o proteínas en la orina, con grados variables de insuficiencia renal. Glomerulonefritis (GN) se refiere a las GP con componente inflamatorio.
Clasificación. Desde el punto de vista clínico, cursan con diversas alteraciones, denominadas síndrome nefrítico, síndrome nefrótico, alteraciones del sedimento urinario y hematuria macroscópica recidivante. Además, pueden producir insuficiencia renal aguda, rápidamente progresiva y crónica. No obstante, estas denominaciones clínicas son insuficientes para caracterizar con precisión estos procesos, por lo que ha sido fundamental la introducción de criterios anatomopatológicos en su clasificación.
Etiopatogenia. Ciertas GN se producen en el contexto de enfermedades sistémicas (secundarias), mientras que en otras las lesiones renales se desarrollan de forma aislada (primarias). (1)
Muchas alteraciones del glomérulo se dana nivel de alguno de sus componentes esperficamente.
Los glomérulos están formados por una red de capilares anastomosados, revestidos por un epitelio visceral (podocitos). Las capas que se interponen entre la luz capilar y el espacio urrinario se denominan barrera de filtración, y son las siguientes:
Los glomérulos están formados por una red de capilares anastomosados, revestidos por un epitelio visceral (podocitos). Las capas que se interponen entre la luz capilar y el espacio urrinario se denominan barrera de filtración, y son las siguientes:
- Endotelio capilar fenestrado
- Membrana basal glomerular (MBG) formado por: colágeno tipo IV, laminina, proteoglucanos polianiónicos (heparán sulfato), fibronectina, entactina, etc.
- Lámina rara interna
- Lámina densa (electrodensa)
- Lámina rara externa
- Epitelio visceral: Podocitos. Células con prolongaciones llamadas pedicelos que recubren la MBG. Entre los pedicelos quedan espacios llamados ranuras de filtración.
El ovillo glomerular se apoya sobre células mesangiales, que sintetizan la matriz que las rodea (matriz mesanguial). Éstas células tienen función contráctil, fagocítica y de secreción de mediadores.


ALTERACIONES HISTOLÓGICAS:
- Hipercelularidad:
- Proliferación celular (mesangiales o endoteliales)
- Infiltrado leucocitario (neutrófilos y macrófagos, y a veces linfocitos)
- Formación de semilunas
- Engrosamiento de la membrana basal glomerular (MBG)
- Hialinosis (material eosinófilo amorfo extracelular, correspondiente a proteínas plasmáticas que han pasado desde la circulación hacia las estructuras glomerulares) y esclerosis (acumulaciones de la matriz de colágeno extracelular). Ambas pueden terminar obliterando la luz capilar.
- Fibrosis
- Trombosis
- Necrosis
PATOGENIADE LAS GP:
1) Mediada por Ac
- Formación de inmunocomplejos in situ:
- Ag fijos, propios del riñon (GN por anti-MBG) Patrón lineal
- Ag implantados: endógenos (LES, tumorales) o exógenos (microbianos, drogas) Patrón granular.
- Depósito de inmunocomplejos circulantes (formados por Ag endógenos o exógenos). Patrón granular.
- Anticuerpos (lesión citotóxica): Mesangio (lisis y proliferación), endotelio (trombosis), epitelial visceral.
2) Inmunidad mediada por células: lesión mediada por macrófagos y LT
3) Activación del complemento por la vía alternativa: en GNMP tipo II y algunas GN proliferativa.
4) Lesión del podocito: Por Ac, drogas, citocinas, mutaciones del componente del diafragma. Morfológicamente se ven cambios en la célula epitelial: borramiento de los podocitos, vacuolización, retracción y desprendimiento de las células de la MBG y funcionalmente se ve proteinuria.
5) Pérdida de nefronas: la reducción del FG a menos del 30-50% de lo normal lesiona las nefronas
remanentes progresando a la insuficiencia terminal por glomeruloesclerosis focal y segmentaria (por hipertrofia e hipertensión glomerular)
MEDIADORES DE LA LESIÓN GLOMERULAR

- Glomeruloesclerosis focal y segmentaria (GEFS)
- Fibrosis tubulointersticial

TIPOS DE GLOMÉRULOPATÍAS Y CUADROS CLÍNICOS


Las distintas glomerulopatías dan distintos cuadros clinicos, se representa con un + los más frecuentes para cada GP.
CLÍNICA:
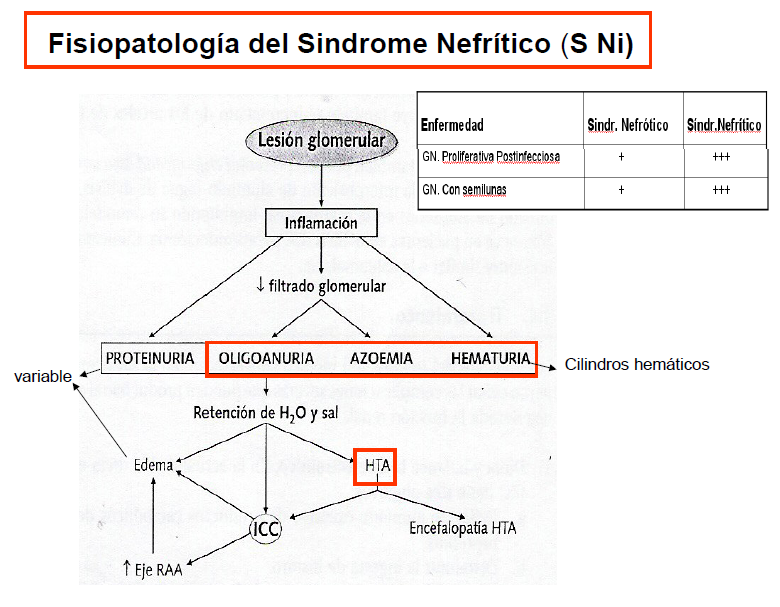
Síndrome Nefrítico:
- Hematuria
- Cilindros hemáticos en orina
- Azotemia
- Oliguria
- HT leve/ moderada
- Puede haber proteinuria y edemas leves
Síndrome Nefrótico:
- Proteinuria masiva, mayor a 3.5 gr/día
- Hipoalbuminemia (menor a 3 gr/dl)
- Hiperlipidemia y lipiduria
- Edema generalizado
El sme nefrótico puede deberse a causas primarias del riñon (las más frecuentes son: enfermedad de cambios mínimos, glomerulopatía membranosa y glomeruloesclerosis focal y segmentaria) o causas sistémicas (como diabetes, amiloidosis y LES).

A continuación se describirá cada una de las glomerulopatías
1- Postinfecciosa
- Postestreptocócica (más frecuente)
- NO estreptocócicas: bacterianas (endocarditis estafilocócica, neumonía neumocócica), virales (HB, HC, parotiditis, HIV, varicela, VEB) o parasitarias (malaria, toxoplasmosis)
2- Enfermedades multisistémicas (secundaria): LES
Postestreptocócica
Etiología: Infección por EGA (estreptococo grupo A) beta hemolítico, cepas 14, 4 y 1 (nefritógenas)
Patogenia: GN mediada por inmunocomplejos (circulantes o Ag plantados)
Los Ag. específicos asociados a la lesión glomerular se cree que son:
- Ag catiónicos: Receptor de plasmina estreptocócica asociado a nefritis (NAPIr)
- Exotoxina Piógena Estreptocócica B (SpeB) y su precursor cimógeno (zSpeB). Ambos actúan como receptor de plasmina.
Morfología:
MO:
- Glomérulos aumentados de tamaño, hipercelulares:
- Proliferación de células endoteliales y mesangiales
- Infiltración leucocitaria (neutrófilos, monocitos)
- Formación de semilunas (solo en casos graves)
- Puede haber edema e inflamación tubulointersticial
IF (inmunofluorescencia)
Patrón granular: Depósito de IgG, IgM y C3 en mesangio y MBG
ME:
Jorobas subepiteliales
Clínica:
Se produce1 a 4 semanas después de una faringitis o impétigo por EGA
Comienza con malesatar, náuseas, fiebre, oliguria y hematuria (orina color coca-cola).
Clínica de sme. nefrítico.
Variantes:
Niños (más frecuente) de 6 a 10 años. Evolución: cura el 95 % de los casos.
Adultos: Epidémico (buena evolución). Esporádico 60 % recupera, 40% evoluciona a GN crónica o GNRP.
Clínica de sme. nefrítico.
Variantes:
Niños (más frecuente) de 6 a 10 años. Evolución: cura el 95 % de los casos.
Adultos: Epidémico (buena evolución). Esporádico 60 % recupera, 40% evoluciona a GN crónica o GNRP.
GLOMERULONEFRITIS RÁPIDAMENTE PROGRESIVA (CON SEMILUNAS)
Etiología: Variada
Patogenia:
- GNRP tipo I: formación de inmunocompleños In situ, por Ag endógenos de la MBG.
- GNRP tipo II: Depósito de complejos inmunitarios. Complicación de otras nefritis.
-GNRP tipo III o pautiinmunitario: Idiopática (50%), secundaria a vasculitis sistémicas (granulomatosis de Wegener, Poliangeítis microscópica), asociados a ANCA.
- GNRP tipo I: inducida por Ac anti-MBG: depósito de IgG y C3 en la MBG (patrón lineal). El depósito puede limitarse al riñon, o los Ac pueden hacer una reacción cruzada con los la membrana basal de los alvéolos pulmonares, en tal caso (que afecta riñon + pulmón) configura el Sme. de Goodpasture. Ag específico en Goodpasture: péptido de la cadena alfa del colágeno tipo IV de la MBG.
- GNRP tipo II: Complicación de otras nefritis mediadas por depósito de inmunocomplejos: postinfecciosas, nefritis lúpica, Púrpura de Schonlein (nefropatía IgA), etc
- GNRP tipo III: Ausencia de complejos inmunes y Ac anti-MBG. Hay ANCA circulantes aunque no se han determinado con causa de la enfermedad. Pueden ser parte de una vasculitis que afecta al riñon, o bien idiopática (sin manifestaciones de vasculitis)
Morfología:
MO: Semilunas: Proliferación epitelial (extracapilar) difusa + macrofagos y monocitos en el espacio urinario. Las semilunas pueden obliterar ell espacio urinario y comprimir el ovillo capilar. También puede haber fuga de fibrinógeno hacia el espacio urinario, que se convierte en fibrina y contribuye a la formación de semilunas.
IF: IgG con patrón lineal en la MBG y C3 en GNRP tipo I. Patrón granular en GNRP tipo II. Ausencia de ambos en GNRP tipo III
ME: Roturas envidentes en la MBG en los tres casos de GNRP. Es la lesión más grave, que permite a los leucocitos, proteínas y mediadore de la inflamación llegar al espacio urinario.
Con el tiempo las semilunas terminan en esclerosis.
Clínica:
- Hematuria con cilindros hemáticos en orina
- Generalmente manifestaciones de sme. nefrítico.
- Generalmente manifestaciones de sme. nefrítico.
- Proteinuria moderada, que en ocaciones puede superar el rango nefrótico, con edema e HT variables
Ráída evolución a Insuficiencia Renal Aguda en semanas o meses.
En Sme de Goodpasture, cursa con hemoptisis y/o hemorragia pulmonar, potencialmente mortal.

NEFROPATÍA MEMBRANOSA
Enfermedad crónica, autoinmunitaria
Enfermedad crónica, autoinmunitaria
Etiología:
- Idiopáticas: 85%
- Secundarias:
- Fármacos: AINE, penicilamina, captopril, oro.
- Tumores malignos subyacentes: carcinoma de pulmón y colon, melanoma.
- LES: El 10-15 % de las glomerulopatías que provoca
- Infecciones: HB, HC, sífilis, malaria, etc
- Enf autoinmunes: glomerulopatía secundaria. ej. tiroiditis
Patogenia: Mediada por complejos inmunes.
- En las secundarias, depende de la enfermedad que lo provoque:
- Depósito de Ag- AC: LES
- Ag. exógenos: HB, Treponema
- Ag. endógenos no renales: Nefropatía membranosa neonatal
- En los casos idiopáticos se desconoce el antígeno desencadenante
Los neutrófilos, monocitos y plaquetas nos participan en el mecanismo de lesión de la membrana. Se cree que el principal causante de la lesión es el complemento. El complejo C5b- C9 activaría las células epiteliales glomerulares y mesangiales, induciendo la liberación de proteasas y oxidantes, que causarían la lesión capilar y el aumento de la pérdida de proteínas.
Morfología:
MO: Engrosamiento difuso uniforme de la pared capilar glomerular.
ME: El engrosamiento se debe a depósitos densos irregulares de complejos inmunitarios entre la MBG y las células epiteliales subyacentes con afectación de los podocitos.
Se forman espículas que hacen protrusión desde la MBG que se ven mejor con tinciones de plata (que tiñen MBG pero no los depósitos). Con el tiempo las espículas se engruesan y forman cúpulas. Al final, se cierran sobre los depósitos inmunes. IF: Los depósitos tienen Ig y complemento
Con el avance de la enfermedad se puede producir esclerosis

Clínica
Es la causa más común de sme. nefrótico en el adulto.
Proteinuria no selectiva (además de albúmina hay pérdida de Ig) y no responde a corticoides.
La esclerosis hallada en una biopsia renal es signo de mal pronóstico.
ENFERMEDAD DE CAMBIOS MÍNIMOS
Etiología: Se asocia con trastornos inmunitarios
Patogenia: Una disfunción inmunitaria daría lugar a la elaboración de una citocina que dañaría las células epiteliales viscerales y causaría proteinuria. Éste daño estaría asociado a la pérdida de polianiones de la barrera de filtración, disminuyendo la selectividad y causando la proteinuria.
Morfología:
MO: Normal
ME: MBG normal sin depósito electrodenso. La lesión de ve en las células epiteliales viscerales, con borramiento difuso de los podocitos, que son reemplazados por citoplasma con vacuolización, inflamación e hiperplasia de las vellosidades.
Las células de los túbulos proximales contienen lípidos y proteínas, por la reabsorción de lipoproteínas que atravisan la barrera de filtración lesionada. Por esto antes se le llamaba "Nefrosis lipoidea"
IF: Normal

Clínica:
Principal causa de sme nefrótico en niños (2-6 años)
Proteinuria selectiva (principalmente albúmina)
Responde al tratamiento con corticoides.
Buen pronóstico
GLOMERULOESCLEROSIS FOCAL Y SEGMENTARIA (GEFS)
Focal: Algunos glomérulos, no todos
Segmentario: Sólo una parte del glomérulo
Etiología y patogenia:
Primarias: Idiopática. Podría deberse a un estado más avanzado de la enfermedad de cambios mínimos. O que podria haber mutación de prot de la hendidura de filtración, entre los pedicelos: NPHS1 que codifica para nefrina, NPHS2 que codifica para podocina, gen de alfa actinina 4 y TRPC6.
Secundarias:
- Infeccion por HIV, nefropatía por heroína, etc
- Secundaria a la cicatrización de lesiones necrosantes por otras GN (ej nefropatía por IgA.)
- Adaptación por pérdida de tejido renal (ver mecanismos de progresión más arriba)
- Enfermedades hereditarias infrecuentes
Se produce daño epitelial visceral, por citocinas o defectos en la hendidura de filtración
Morfología
MO: En los segmentos escleróticos hay asas capilares colapsadas, aumento de la matriz y depósito segmentario de proteínas siguiendo la red capilar (hialinosis).
Puede haber gotículas de lípidos y células espumosas.
Los glomérulos sin esclerosis se ven normales al MO
ME: Áreas escleróticas y no escleróticas con borramiento difuso de los podicitos, desprendimiento focal de las células epiteliales y denudación de la MBG subyacente.
IF: IgM y C3 en áreas escleróticas y/o mesangio.
Con el tiempo se produce una esclerosis total (global) de los glomérulos, con atrofia tubular y fibrosis intersticial.
En la nefropatía por HIV, es característica la proliferación e hipertrofia de las células epiteliales viscerales, con lesión tubular y formación de microquistes. Tiene mal pronóstico.
Clínica:
- Hematuria, disminución del FG e HT
- Proteinuria generalmente no selectiva
- Respuesta a corticoides insuficiente
- Al menos el 50% progresa a nefropatía terminal antes de los 10 años
GLOMERULONEFRITIS MEMBRANOPROLIFERATIVA (GNMP)
Se caracteriza por alteraciones de la MBG, proliferación de células del glomérulo e infiltrado leucocitario.
También llamada GN mesangiocapilar, ya que la proliferación se da en el mesangio y las asas capilares.
Etiología y patogenia:
- Primaria: idiopática
- Tipo I: Podría ser causada por Ag postinfección, que actúan como Ag plantados o complejos depositados desde la circulación. Activación del complemento (vía clásica y alternativa)
- Tipo II o enfermedad de los depósitos densos. Activación de la vía alternativa del complemento. Tienen disminuídos los valores séricos de C3, factor B y properdina, mediadores de la vía alternativa. En cambio C1 y C4 que participan de la vía clásica se encuentran normales. El 70% tiene autoanticuerpo "factor nefrítico C3 (C3NeF)".
El complemento se puede activar de tres maneras:
Vía Clásica, necesita el antígeno unido a un anticuerpo + C1 + C2 + C4 para activarse.
Vía alternativa, se puede activar sólo con el contacto con endotoxinas y polisacáridos bacterianos + factores B y D.
Vía de las lectinas, necesita que la lectina de unión a manosa soluble en el suero se una a las manosas de la pared de las bacterias.
Las tres vias resultan en la formación de C3 convertasa, que activa C3, a partir de éste punto común, se consumen otras proteínas del complemento con el resultado de la formación del compelejo lítico de ataque C56789, que forma un poro en las membranas. En activación de las distintas proteínas del complemento se produce una amplificación y liberación de intermediarios proinflamatorios (quimiocinas y anafilatoxinas)
La vía alternativa está regulada por los factores I y H que degradan c3 convertasa y la properdina que la estabilizan. En condiciones normales hay un equilibrio entre ambos mecanismos. En la enfermedad de depósitos densos, el autoanticuerpo C3NeF se une a C3 convertasa y la estabiliza, dando una activación continua del complemento que consume los precursores inactivos de la via alternativa. Es por eso que C3, el factor B y la properdina se encuentran disminuídos
- Trastornos crónicos por complejos inmunitarios (LES, HB, HC, etc)
- Deficiencia de alfa1-AT
- LLC y linfoma
- Deficiencia de proteínas reguladoras del complemento
No se conoce el mecanismo del depósito de complejos inmunes en los últimos tres.
Morfología:
MO: Glomérulos grandes e hipercelulares. La hipercelularidad es por proliferación de células mesangiales y endotelio capilar, con leucocitos infiltrantes.
Puede haber semilunas.
Glomérulos con aspecto "lobulado"
MBG engrosada
Asas capilares: Paredes en "doble contorno" o en "vías del tren", por desdoblamiento o duplicación de la membrana basal
Tipo I:
ME: depósitos electrodensos subendoteliales delimitados
IF: C3, IgG, C1 y C4 (muestra activación de la vía clásica por complejos inmunitarios y alternativa)
Tipo II
ME: Depósito de material denso en la MBG
IF: C3 en MBG y mesangio (anillos mesangiales).
Clínica:
Síndrome nefrítico/nefrótico.
Adolescencia y adultos jóvenes
El 50 % desarrolla IRC antes de los 10 años
No se ha demostrado tratamiento eficaz.
Recurrente en transplantados
RESUMEN:

GLOMERULOPATÍAS SECUNDARIAS:
- Nefropatía diabética
- Nefritis Lúpica
NEFROPATÍA DIABÉTICA
La diabetes mellitus es una de las principales causas de insuficiencia renal terminal.
Los cambios renales en ambos tipos de diabetes son similares morfológicamente y en cuanto a su fisiopatología. Se asocia con sme. nefrótico.
La patogénesis de la ND no está completamente entendida. La glicosilación no
enzimática de proteínas parece ser uno de los principales mecanismos de lesión
glomerular; los productos terminales de la glicosilación avanzada pueden unirse a
grupos amino de otras proteínas, su acumulación en el colágeno y otras proteínas
de la matriz puede disminuir la adhesión y replicación de células endoteliales,
vasoconstricción por células mesangiales e incrementar la adhesión de lipoproteínas
y complejos inmunes a monocitos y macrófagos. Los factores hemodinámicos que
llevan a hiperfiltración glomerular es otro mecanismo de daño renal.
Morfología: Incremento de la matriz mesangial y engrosamiento de membranas basales y de la íntima de arterias y arteriolas.
engrosamiento de las paredes capilares, estos cambios son más evidentes con icroscopía
electrónica. Al progresar las lesiones hay aumento, también, de la celularidad mesangial; este incremento llega hasta la formación de nódulos en el penacho. Los nódulos son de tamaño variable en un mismo glomérulo y afectan de una manera heterogénea los diferentes glomérulos (glomeruloesclerosis nodular diabética). Los nódulos son conocidos como nódulo de Kimmelstiel-Wilson. Son esféricos, eosinofílicos, con un área central acelular y pueden esta rodeados por un anillo celular. Tiñen azul o verde con el tricrómico y son positivos con las tinciones de PAS y plata-metenamina. Los nódulos son de dos tipos morfológica y patogénicamente: unos son pequeños, mesangiales (o intercapilares) y los otros son grandes con aspecto laminado en la tinción de plata. Los grandes se formarían a partir de capilares glomerulares dilatados (microaneurismas, >35 micras de diámetro) que generarían lesión en la pared capilar, trombosis y proliferación de matriz colágena (4). Los pequeños se ven como una acentuación redondeada de la matriz esangial y tiñen más intensamente con el PAS que los nódulos grandes y laminados. La presencia de estos nódulos: grandes y pequeños, algunos laminados, con tamaño y distribución variable en el glomérulo y entre los glomérulos, es "virtualmente patognomónica" de ND, Los nódulos vistos en la enfermedad por depósitos de cadenas ligeras pueden ser parecidos, pero más homogéneos en tamaño y distribución y tiñen más débilmente, o son negativos, con la tinción de plata; los nódulos de la amiloidosis no tiñen con la plata y son positivos para rojo congo. La IF también ayuda en el diagnóstico diferencial.
En ND los glomérulos presentan esclerosis de la matriz mesangial intercapilar,
con aumento progresivo del engrosamiento de paredes capilares y posterior evolución a
glomeruloesclerosis global .
Clínica:
El hallazgo más temprano de la afectación renal es microalbuminuria que progresa
gradualmente hasta proteinuria, usualmente alrededor de los 15 años del inicio de la enfermedad. La ND se desarrolla en aproximadamente un 30% de pacientes diabéticos. La hipertensión arterial sistémica y el tabaco incrementan el riesgo de nefropatía en diabéticos.
Las lesiones renales crónicas evolucionan progresivamente; el daño del tejido renal es complejo y compromete glomérulos, vasos (como en cualquier otro órgano o sistema), intersticio y túbulos. Al progresar las lesiones crónicas se disminuye la reserva funcional y al pasar el límite crítico de lesión parenquimatosa comienzan a detectarse grados variables de insuficiencia renal que continuará progresando hasta la insuficiencia renal crónica terminal.
El hallazgo más temprano de la afectación renal es microalbuminuria que progresa
gradualmente hasta proteinuria, usualmente alrededor de los 15 años del inicio de la enfermedad. La ND se desarrolla en aproximadamente un 30% de pacientes diabéticos. La hipertensión arterial sistémica y el tabaco incrementan el riesgo de nefropatía en diabéticos.
Las lesiones renales crónicas evolucionan progresivamente; el daño del tejido renal es complejo y compromete glomérulos, vasos (como en cualquier otro órgano o sistema), intersticio y túbulos. Al progresar las lesiones crónicas se disminuye la reserva funcional y al pasar el límite crítico de lesión parenquimatosa comienzan a detectarse grados variables de insuficiencia renal que continuará progresando hasta la insuficiencia renal crónica terminal.
Morfología:
- Engrosamiento de la MBG
- Esclerosis mesangial difusa
- Glomeruloesclerosis nodular (enf. de kimmelstiel- Wilson)
Clínica
- Microalbuminemia 30 a 300 mg/día (aumento subclínico de la excreción urinaria de albúmina
- Proteinuria de rango no nefrótico
- Insuficiencia terminal

Nódulos de Kimmelstiel- Wilson
NEFRITIS LÚPICA

Otra forma de agruparlas:
Glomerulopatías primarias (ausencia de enfermedad sistémica)
a)Proliferativas.
1. Endotelio capilar. GN proliferativa difusa Aguda. (postestreptocícica + frec.)
2. Mesangio proliferativa. Crónicas
3. Membrano proliferativa. Crónicas
4. Proliferación epitelial. Glomerulonefritis Rápidamente Progresiva (GNRP), con semilunas. Subagudas
a)Proliferativas.
1. Endotelio capilar. GN proliferativa difusa Aguda. (postestreptocícica + frec.)
2. Mesangio proliferativa. Crónicas
3. Membrano proliferativa. Crónicas
4. Proliferación epitelial. Glomerulonefritis Rápidamente Progresiva (GNRP), con semilunas. Subagudas
b) NO proliferativas.
1. Cambios mínimos
2. Esclerosis focal y segmentaria
3. Membranosa
4. Mesangial leve
Glomerulopatías Secundarias (por enfermedad sistémica)
a) Síndrome nefrótico
1. Amiloidosis
2. Diabetes mellitus
3. Lupus eritematoso sistémico
4. Mieloma múltiple
b) Síndrome nefrítico
1. Glomerulonefritis post-estreptocóccica
2. Glomerulonefritis lúpica
3. Glomerulonefritis membrano-proliferativa
4. Glomerulonefritis crioglobulinémica
Otras:
1. Vasculitis: Schönlein Henoch.
Panarteritis nodosa. Wegener
2. Goodpasture
3. Púrpura trombocitopénica trombótica-
Síndrome urémico hemolítico
4. Mesangio-proliferativa
(3)
Bibliografía
(1) Glomerulopatías. Concepto. Clasificación. Etiopatogenia; Patricia Martínez Miguel, D. Rodríguez Puyol; Medicine, ISSN 0304-5412, Serie 9, Nº. 80, 2007, págs. 5125-5130
(2) Robbins y Cotran. Patología estructural y funcional. Octava edición. Ed. Elsevier Saunders.
(3) Revista de la Sociedad de Medicina Interna de Buenos Aires. Las Glomerulopatías. Enfoque Clínico-patológico; Dr. Carmelo Pietrángelo (Doctor en Medicina de la UBA. Docente Autorizado de la Facultad de Medicina de la UBA.Jefe de Unidad de Internación del Hospital de Clínicas Jose de San Martín)
(4) Stout LC, et al., Hum Pathol; 24:77-89, 1993
(5) Inmunología celular y molecular; Abul K. Abbas, Andrew H. H. Lichtman, Shiv Pillai; Séptima edicion. Ed. Elsevier Saunders.
(6) Argente- Álvarez. Semiología médica, fisiopatología, semiotecnia y propedéutica. Ed. Panamericana









